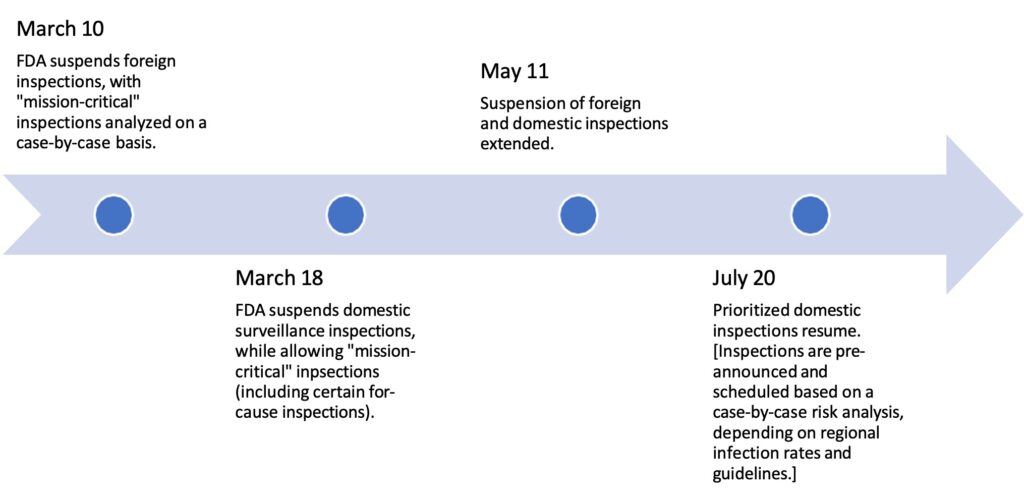
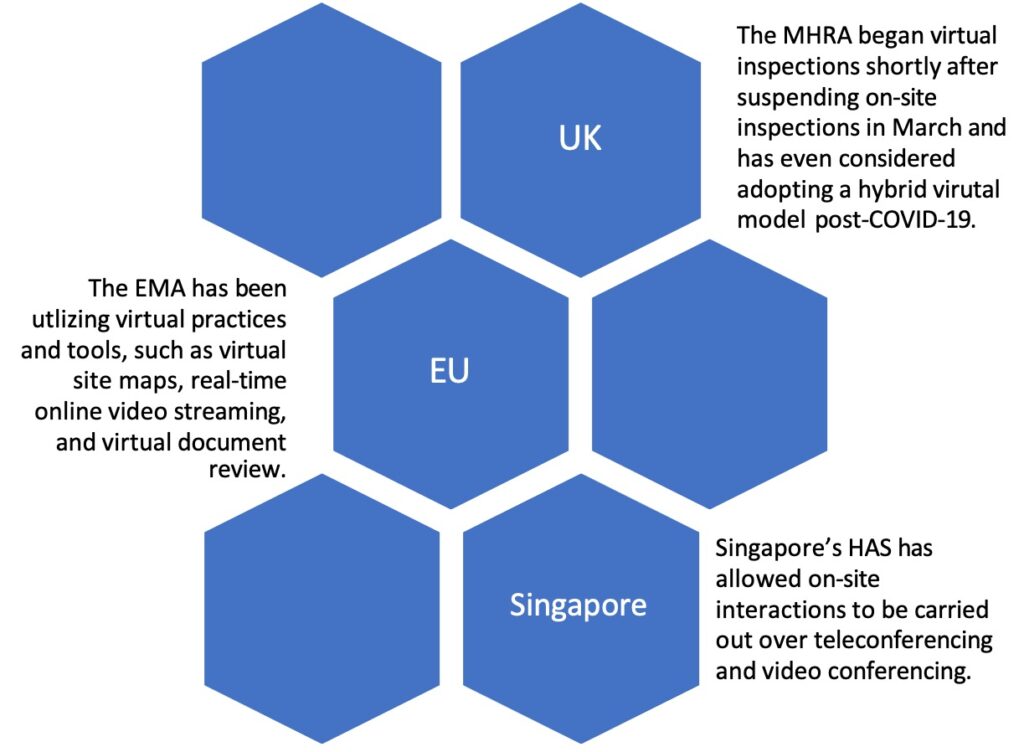
Greenleaf Regulatory Landscape Series
For nearly two decades, the Food and Drug Administration (FDA or the Agency) has supported the development of innovative manufacturing technologies that modernize quality management systems and provide greater quality assurance across medical product supply chains. Evidence of the FDA’s commitment to the development and implementation of this technology appeared most recently in January 2021 with the creation of a new collaboration between the FDA and the National Institute of Standards and Technology (NIST), under a Memorandum of Understanding (MOU), aiming to increase U.S. supply chain resilience and advanced domestic manufacturing by adopting innovative technologies, such as continuous manufacturing processes, as well as artificial intelligence and machine learning. The purpose of this effort is to mitigate risks of supply chain disruptions leading to product shortages, a concern that has grown more problematic under an increasingly globalized approach to medical product production.
Since the onset of the coronavirus pandemic (COVID-19) in Spring 2020, similar efforts to support uptake of advanced manufacturing technologies have been met with a greater sense of urgency across other parts of the federal government as well. Consistent with these efforts, actions to on-shore and increase domestic manufacturing capacity and modernize medical product manufacturing have been bolstered by pandemic-related legislation and executive orders alike.
Federal Initiatives Related to Medical Product Supply Chains During COVID-19
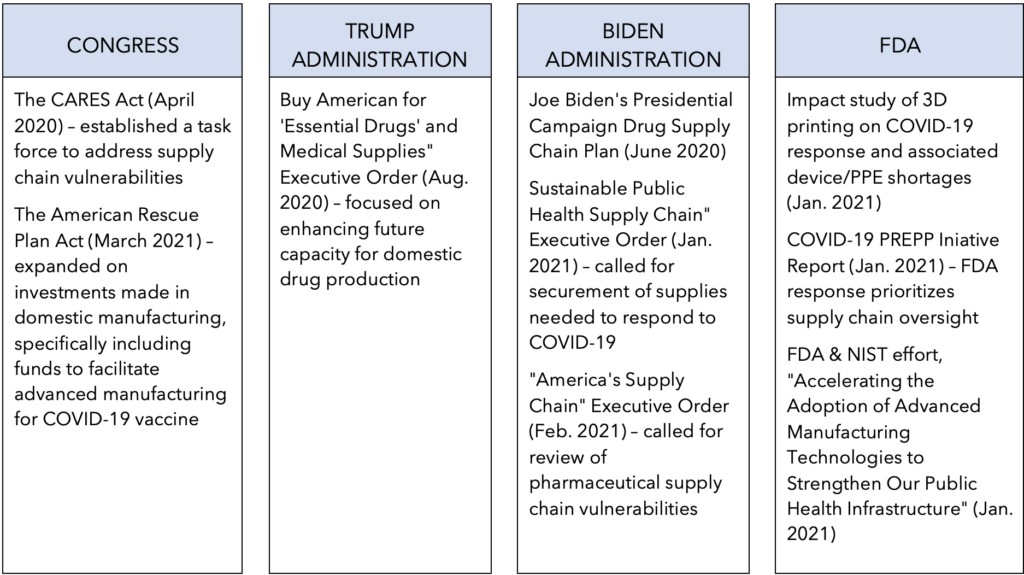
Even though acute awareness of the potential benefits of advanced manufacturing resurfaced during the pandemic, the origins of the FDA’s pursuit of modern quality systems through advanced manufacturing began in August 2002 with the launch of its “Pharmaceutical Current Good Manufacturing Practices (cGMPs) for the 21st Century” initiative. This initiative evaluated the Agency’s pharmaceutical regulatory programs and released a final report in September 2004, introducing a new risk-based quality assessment system that would replace the chemistry, manufacturing, and controls (CMC) review process, encouraging implementation of process analytical technologies, and framing innovative technologies as essential components of a modern quality system. Later that same year, the FDA issued final guidance for industry, “Process Analytical Technology (PAT) and a Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance,” establishing a regulatory framework intended to support more innovation and quality modernization in pharmaceutical production.
Part of the Agency’s intention in its initial push for adopting advanced manufacturing technologies stemmed from concerns about quality management deficits associated with conventional batch manufacturing processes. The decades-old batch manufacturing model consists of frequent testing, storage, and shipping across regions, making it time-sensitive and more prone to product contamination. In addition, the batch manufacturing model was linked to reactive and wasteful discarding of final drug products due to quality issues. Instead, the FDA envisioned transitioning to a risk-based, quality management framework involving a more controlled and efficient pharmaceutical production regime.
Benefits of a Continuous Manufacturing System Versus
a Batch Manufacturing System
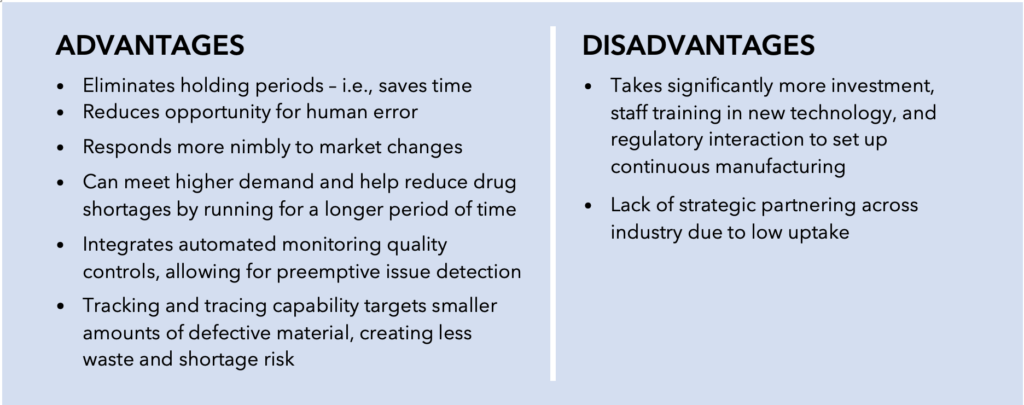
Pursuant to this vision, the FDA supported implementation of advanced manufacturing technologies, such as continuous manufacturing, by establishing platforms for engagement between the Agency and companies interested in producing products using innovative technologies. These platforms involve CDER’s Emerging Technology Program (ETP), CBER’s Advanced Technology Teams (CATT), and CDRH’s Case for Quality initiative. Additional final guidance on “Advancement of Emerging Technology Applications for Pharmaceutical Innovation and Modernization” continued to support companies seeking to adopt advanced manufacturing technologies. Under this regime, nine drug products manufactured with advanced technologies, one of which uses biomanufacturing processes, have been approved by the FDA – three of these products were approved in 2020 according to CDER’s Office of Pharmaceutical Quality (OPQ) Annual Report.
The National Academies of Sciences, Engineering, and Medicine (NASEM) held a workshop in June 2020 on barriers that hinder pharmaceutical manufacturing innovation, finding that external, regulatory challenges “loom large.” At this workshop, CDER-OPQ Director Mike Kopcha, Ph.D., distilled what he saw as regulatory barriers to the adoption of advanced manufacturing, including the need to fit new technologies into existing regulatory frameworks and the need for global regulatory harmonization. That is, because the current regulatory framework is “based in offline testing of batch processes, regulatory requirements do not currently translate well into new manufacturing technologies that allow for varied batch sizes, inline analytics, and higher-fidelity methods for detecting batch-to-batch variation.”
While the FDA has done much in the way of encouraging industry implementation of innovative and emerging technologies, barriers to achieving more substantial implementation persist. Earlier this year, NASEM released a report highlighting gaps where future guidance and clarity would be helpful in mounting identified regulatory barriers. These include:
- Consideration of more fluid and targeted guidance that is shorter and published promptly to allow for industry comment.
- Greater focus on underlying technology, as opposed to individual product approvals.
- Creation of new mechanisms and pilot programs for incorporation of industry input and collaboration.
- Expanded scope of the ETP to create greater opportunities for interested companies to engage.
With that said, the FDA is working to finalize its February 2019 draft guidance on “Quality Considerations for Continuous Manufacturing,” in which it has the opportunity to respond to gaps identified by NASEM, as well as others. With Acting Commissioner Janet Woodcock, M.D., at the helm, the FDA’s commitment towards advanced manufacturing technologies is likely to remain a top priority – Dr. Woodcock has long championed the increased adoption of advanced manufacturing technologies throughout her FDA career. Even if a permanent nominee other than Dr. Woodcock were to take her place as FDA Commissioner, advanced manufacturing is still seen as a key element to modern, risk-based quality systems and part of secure and efficient supply chains. Therefore, the FDA’s support of the adoption of advanced manufacturing will continue. Additionally, as previously noted, efforts to strengthen medical product supply chains by focusing on advanced technologies and re-focusing on U.S.-based manufacturing has enjoyed recent bipartisan support, in large part due to lessons learned from the pandemic. Thus, although industry adoption has been slow, advanced manufacturing technologies are expanding in scope and will likely become more of a norm in a post-COVID-19 world.
Resources
- FDA & NIST MOU, “Accelerating the Adoption of Advanced Manufacturing Technologies to Strengthen Our Public Health Infrastructure” (January 2021)
- Trump Executive Order, “Buy American for ‘Essential Drugs’ and Medical Supplies” (August 2020)
- Biden Presidential Campaign, “Rebuild U.S. Supply Chains and Ensure the U.S. Does Not Face Future Shortages of Critical Equipment” (June 2020)
- Biden Executive Order, “Sustainable Public Health Supply Chain” (January 2021)
- Biden Executive Order, “America’s Supply Chains,” (February 2021)
- FDA Initiative, “3D Printing in FDA’s Rapid Response to COVID-19” (November 2020)
- FDA PREPP Initiative, “FDA’s COVID-19 Pandemic Recovery and Preparedness Plan (PREPP) Initiative: Summary Report” (January 2021)
- FDA In Brief, “FDA Provides Update on COVID-19 Pandemic Recovery and Preparedness Plan Initiative” (April 2021)
- FDA Initiative – Final Report, “Pharmaceutical CGMPs for the 21st Century: A Risk-Based Approach” (September 2004)
- FDA Final Guidance, “PAT – A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance” (October 2004)
- FDA Final Guidance, “Advancement of Emerging Technology Applications for Pharmaceutical Innovation and Modernization” (September 2017)
- FDA Draft Guidance, “Quality Considerations for Continuous Manufacturing” (February 2019)
- NASEM Workshop – Proceedings in Brief, “Barriers to Innovations in Pharmaceutical Manufacturing” (September 2020)
- NASEM Report, “Innovations in Pharmaceutical Manufacturing on the Horizon: Technical Challenges, Regulatory Issues, and Recommendations” (2021)