FDA Inspectional Activities During COVID-19
Greenleaf Regulatory Landscape Series
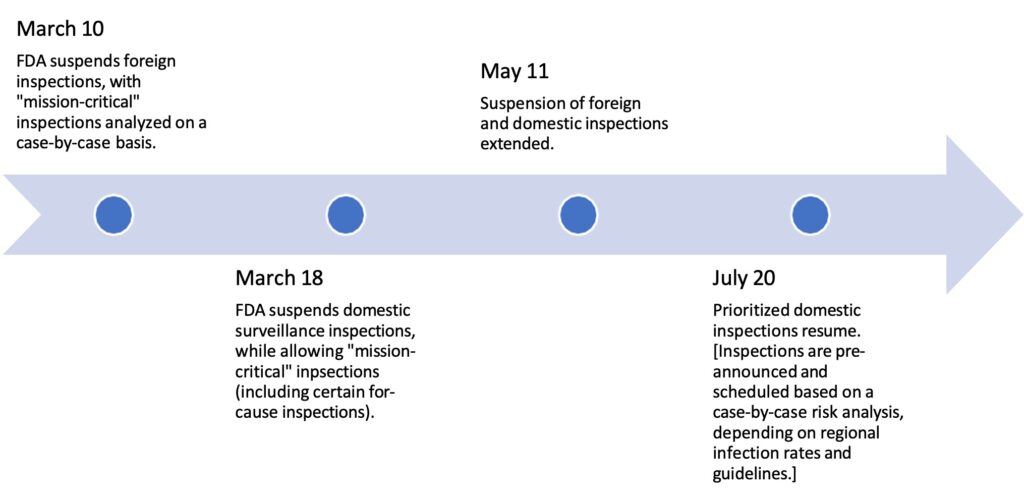
On March 10, 2020, following the global emergence of the novel coronavirus (SARS-CoV- 2) and spread of COVID-19 disease, the Commissioner of the Food and Drug Administration (FDA or the Agency) suspended most foreign inspections of facilities that manufacture FDA- regulated products. The following week, domestic surveillance inspections, if not deemed “mission-critical,” were postponed as well. These policies were extended in subsequent months, with prioritized, pre-announced domestic inspections only resuming in July 2020. As the pandemic progressed, these travel restrictions and on-site limitations led to a growing backlog of regulatory inspections. Regulators responded by undertaking various alternative approaches to inspectional work to ensure the continued supply of quality medicines to patients, and to mitigate the creation of drug shortages.
Timeline of On-Site Inspection Policies at FDA During COVID-19
Under Section 704(a)(4) of the Food, Drug, and Cosmetic Act (FDCA), FDA can “in advance of or in lieu of an inspection” request records or information to determine whether inspection of a drug manufacturing facility is needed. Note: Section 704(a)(4) does not apply to other FDA inspection types (e.g., the Bioresearch Monitoring Program (BIMO), good clinical practice (GCP) inspections, medical device manufacturing facility inspections, etc.). Leveraging this authority, FDA began utilizing 704(a)(4) records requests to enable abbreviated on-site inspections so that the risk of exposure to COVID-19 was otherwise reduced. By August 2020, Elizabeth Miller, Assistant Commissioner for Medical Products and Tobacco Operations at FDA, said that 424 records requests to drug manufacturers had been made during the pandemic – 111 in the pre-approval space and 313 in the post-approval, good manufacturing practice (GMP) space. Miller added that another 123 records requests to biologics manufacturers had been made during the same time frame. Additionally, FDA made use of flexibilities established under the Mutual Recognition Agreement (MRA) for products evaluated by the European Union (EU), as well as information from inspections occurring outside the EU provided by the Pharmaceutical Inspection Co-Operation Scheme (PIC/S).
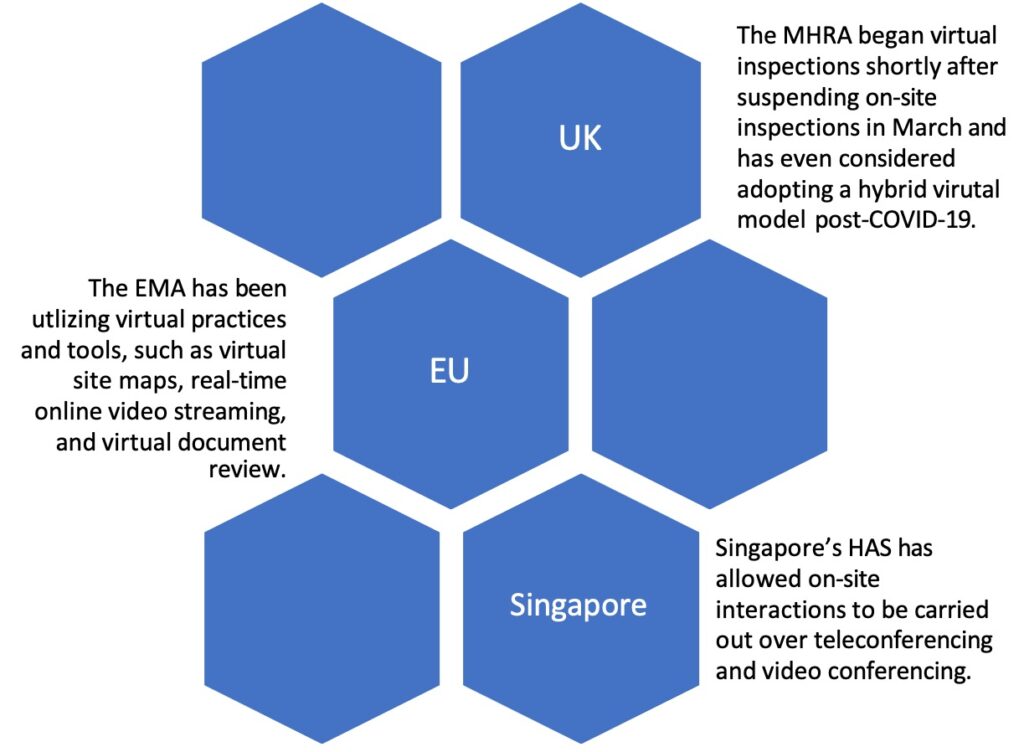
On August 19, 2020, FDA published final guidance, “Manufacturing, Supply Chain, and Drug and Biological Product Inspections During COVID-19 Public Health Emergency Questions and Answers,” which closely followed previously posted guidance from April 2020 and continued “mission-critical” inspections, which had been on-going throughout the pandemic, as well as prioritized, domestic inspections based on a case-by-case risk analysis. Although the new guidance did not indicate whether FDA would begin to lean into virtual technologies as a supplement to 704(a)(4) records requests and/or on-site inspection requirements, foreign regulators demonstrated interest in implementing virtual approaches to their inspectional activities early on in the pandemic. For example, health authorities in the UK, EU, and Singapore piloted various virtual practices and tools as supplements to standard GMP inspections.
Foreign Health Authorities’ Adoption of Virtual Inspectional Activities
In November 2020, speculation grew as to whether FDA would begin conducting some form of virtual GMP assessments or inspections, similar to its foreign counterparts. Brian Hasselbalch, the Deputy Director of the Office of Policy for Pharmaceutical Quality at the Center for Drug Evaluation and Research (CDER), confirmed that guidance “on remote evaluations using interactive video or other types of interactive tools and techniques” is under development. Hasselbalch said that such a document will describe the role these types of interactions are to play in the Agency’s decision making with respect to pending applications, surveillance activities, and possibly even for-cause inspections. He added that manufacturers with pending applications involving facilities, or plans to submit such applications, should begin to equip themselves with necessary streaming technologies.
Despite indications that FDA is planning to utilize virtual technologies in some form, it is unclear what this will mean for inspection requirements under the FDCA. When implementing a virtual approach to inspectional work, the Agency has to work within its Section 704 authority and ensure this authority reaches across various product areas (e.g., drugs, biologics, and medical devices). Importantly, there is a distinction between a virtual assessment, where the Agency would rely on virtual tools to facilitate a 704(a)(4) records request (e.g., by following up on a records request with live streaming of the facility, etc.), and otherwise allowing an actual virtual inspection, where an inspector would present an FDA Form 482 and could issue an FDA Form 483. The approach taken by FDA will depend on whether it interprets a virtual inspection to count as a statutorily required inspection under Section 704 of the FDCA, although the Agency’s stance on this has not been made clear. Irrespective of FDA’s allowance of virtual inspection, or mere virtual assessments as part of 704(a)(4) records request, it seems inevitable that further incorporation of various virtual tools and technologies is approaching, especially as COVID-19 continues to impact regulatory operations and oversight around the world.
Related Sources
- FDA Postpones Foreign Inspections Through April 2020
- FDA Scales Back Domestic Inspections Through April 2020
- FDA Updates Surveillance Inspections During COVID-19 in May 2020
- FDA Prepares for Resumption of Domestic Inspections with New Risk Assessment System in July 2020
- FDA Assesses Over 500 Biopharma Plants Remotely Via Records Review, Refines Process
- Manufacturing, Supply Chain, and Drug and Biological Product Inspections During COVID-19 Q&A Guidance
- MHRA Relies on Remote Drug GMP Inspections
- How the EMA Inspected Thermo Fisher’s Florida Plant Without Leaving Europe
- Singapore HAS Handling of Applications and Conduct of Inspections During COVID-19
- FDA Developing Guidance for Interactive Video GMP Evaluations During COVID-19