Greenleaf Regulatory Landscape Series
Considering the increasing interest in and use of digital health technologies in clinical care and research, it should come as no surprise that several performance goals related to digital health technologies are included in the Food and Drug Administration’s (FDA) user fee legislation1 for the medical device and prescription drug programs for fiscal years 2023 through 2027. Indeed, researchers have found that between 2000 and 2018, the use of digital health tools in clinical trials grew at a compound annual growth rate of 34%.2
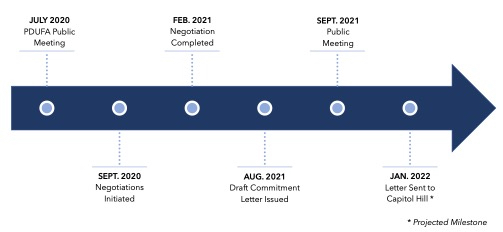
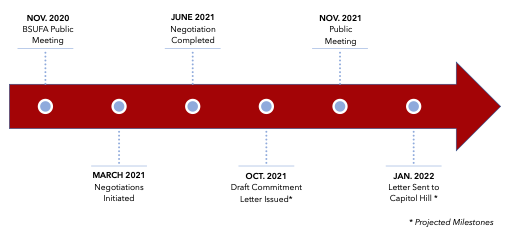
The Medical Device User Fee Amendments (most recently reauthorized as MDUFA IV) and Prescription Drug User Fee Act (most recently reauthorized as PDUFA VI) sunset every five years, unless reauthorized by Congress. This year both User Fee Amendments were reauthorized by Congress on September 29, 2022, one day before MDUFA IV and PDUFA VI’s sunset date. As part of the reauthorization process, the FDA negotiates user fee commitment letters with regulated industry. These commitment letters set forth the performance goals agreed to by the FDA for the next five fiscal years in exchange for outlined user fees to be paid by industry. This landscape describes in detail the digital health commitments included in the user fee commitment letters accompanying the legislation to reauthorize the device and prescription drug user fee programs, MDUFA V3 and PDUFA VII4.
MDUFA V Digital Health Commitments
Several of the FDA’s MDUFA V commitments related to digital health build on agency efforts already in progress. The FDA agreed to “continue to build its digital health expertise and continue working to streamline and align FDA review processes with software lifecycles for digital health products.” More specifically, the FDA agreed to continue to:
- Develop expertise to support the review of premarket submissions that include digitial health technologies like artificial intelligence (AI), virtual reality, and wearables;
- Expand staff understanding of digital health topics and work to ensure the consistent review of digital health-related submissions through training and agency infrastructure;
- Participate in international harmonization efforts related to digital health; and
- Engage with stakeholders through roundtables, informal meetings, and teleconferences to explore regulatory approaches to digital health technologies.
In addition, the FDA committed to finalizing one guidance document and issuing a new draft guidance. The FDA agreed to finalize the draft guidance titled, “Draft Guidance for Industry and FDA Staff: Content of Premarket Submissions for Device Functions” by August 20235 and also to publish a draft guidance document “describing a process to evaluate a predetermined change control plan for digital health devices.”
PDUFA VII Digital Health Commitments
As opposed to the FDA’s broader MDUFA V commitments to continue to build digital health expertise and align FDA review processes, the goals related to digital health in the PDUFA letter are focused more narrowly on developing a regulatory framework to help advance the use of digital health technologies (DHTs) in clinical trials. Highlighting the need for a more robust regulatory framework for DHTs, the FDA states in the commitment letter, “FDA recognizes the potential for DHTs to provide scientific and practical advantages in supporting the assessment of patients by generating information outside of the traditional clinic visit.6 Thus, to encourage the adoption of DHTs in clinical trials to support drug registration, label expansion, and safety monitoring, the FDA has committed to the following goals.
Develop a Regulatory Framework
Similar to the Real-World Evidence (RWE) Framework that the FDA published in December 2018 to outline its proposed regulatory approach to RWE, the FDA has agreed to issue a regulatory framework for digital health technologies by end of the Q2 2023. The document will identify objectives for workshops and demonstration projects, describe methodologies for evaluating DHTs as endpoints, and discuss standardized processes for managing and submitting continuous or extensive datasets.
Establish a DHT Committee
To promote regulatory consistency across the Centers and oversee the development and implementation of the DHT regulatory framework, the FDA has committed to establishing a DHT committee with representatives from both the Center for Drug Evaluation and Research (CDER) and the Center for Biologics Evaluation and Research (CBER). The committee will also be responsible for engaging external stakeholders and gathering intelligence on the current challenges related to DHTs.
Convene Workshop Series on DHTs
By the end of Q2 2023, the FDA will convene a series of five public workshops to gather input from external stakeholders on key issues related to the use of DHTs in regulatory decision-making. As noted in the letter, meetings will likely focus on:
- Using DHTs to increase trial diversity;
- Approaches to DHT validation;
- DHT data processing and analysis techniques; and
- Regulatory acceptance of safety monitoring tools using AI and machine learning.
Launch DHT Demonstration Projects
The FDA will launch a series of demonstration projects designed to inform regulatory policy developments related to DHTs. Projects will likely be designed to further examine validation methods, investigate different approaches to addressing missing data, and inform the use of multi-channel inputs.
Issue Guidance
The FDA has committed to issuing a wide swath of guidances to further develop the DHT regulatory paradigm. By Q1 2023, the FDA will issue draft, revised, or final guidance documents on the following topics:
- Use of DHTs in traditional and decentralized trials;
- Validation of measurement by DHTs;
- Developing novel endpoints and measuring existing endpoints with DHTs;
- Using patients’ own DHTs, such as cellular phones or smart watches;
- DHT usability considerations for patients;
- Detection of safety signals during continuous data acquisition; and
- Data security and confidentiality.
In addition to the abovementioned topics, the FDA has also committed to issuing guidance on regulatory considerations for Prescription Drug Use-Related Software by the end of 2023 and on any other specific topics identified through stakeholder engagement by 2024.
Expand Organizational and Technological Capacity
To accommodate the anticipated wave of regulatory submissions incorporating DHT-derived data, the FDA will have to enhance both its organizational and technological capacities. To develop the technical expertise needed, the FDA will invest in training on digital technologies and on relevant statistical methodologies. The FDA has also committed to establishing a consistent approach to the review of DHT- related submissions across all three Centers. To upgrade the Agency’s technological systems, in FY 2023, the FDA will establish a secure cloud technology to enhance its infrastructure and internal analytics environment to process the large volume of DHT-generated data. The Agency will also work on recommending and implementing data standards to make DHT-derived data more analyzable.

1. See https://www.congress.gov/117/bills/hr6833/BILLS-117hr6833eas.pdf.
2. Mara, C., et al. “Quantifying the Use of Connected Digital Products in Clinical Research,” npj Digital Medicine, 3 (50), 2020, https://www.nature.com/articles/s41746-020-0259-x#Abs1.
3. FDA, “PDUFA Reauthorization Performance Goals and Procedures Fiscal Years 2023 through 2027,” https://www.fda.gov/media/151712/download.
4. FDA, “MDUFA Performance Goals and Procedures, Fiscal Years 2023 through 2027,” https://www.fda.gov/media/158308/download.
5. See https://www.fda.gov/media/153781/download.
6. FDA, “PDUFA Reauthorization Performance Goals and Procedures Fiscal Years 2023 Through 2027,” August 2021, https://www.fda.gov/media/151712/download.