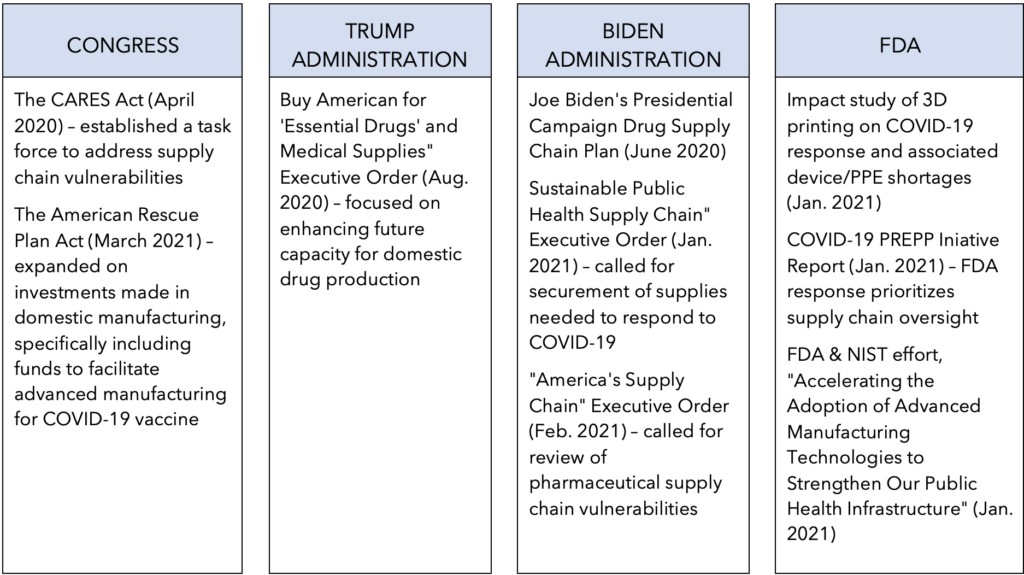
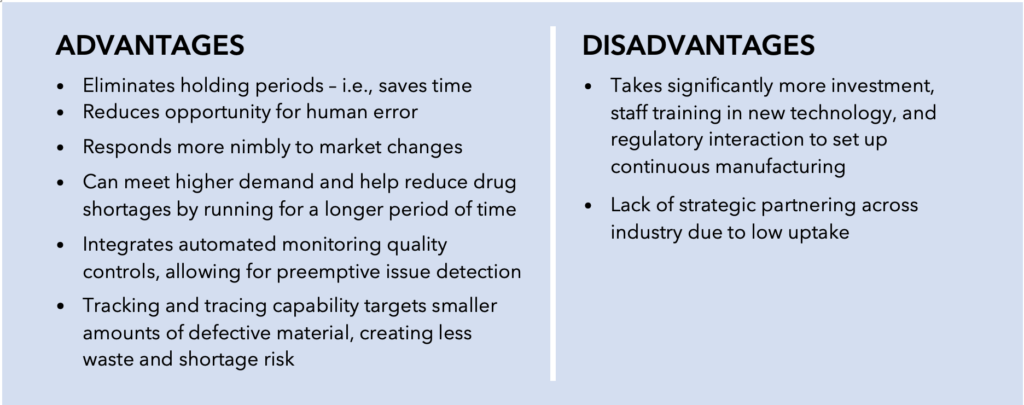
This article was originally published as a guest column in Outsourced Pharma.
Considering the feverish pace of innovation in the field of AI/ML and the inevitable impact this family of technologies has on drug development, an overview of the approaches to AI/ML regulation by the leading medical product regulatory authorities, the FDA and European Medicines Agency (EMA), is timely. Below, we outline the documents and guidances the two regulators have released thus far, comparing and contrasting their areas of focus and concern.
A Comparison of the Definitions of AI and ML
Despite the lack of a universally accepted definition of AI among experts,1 both regulatory agencies have settled on a working definition of AI.
In its definition, FDA acknowledges the breadth and multidisciplinary nature of the field, defining AI as “[a] branch of computer science, statistics, and engineering that uses algorithms or models to perform tasks and exhibit behaviors such as learning, making decisions, and making predictions.”2 Meanwhile, FDA identifies ML as a subset of AI that allows “[m]odels to be developed by ML training algorithms through analysis of data, without being explicitly programmed.”3
EMA, however, takes a more mechanistic approach, defining AI as “systems displaying intelligent behavior by analyzing data and taking actions with some degree of autonomy to achieve specific goals.”4 Meanwhile, EMA’s definition of ML — “models [that] are trained from data without explicit programming” — mirrors FDA’s ML definition.
The FDA’s Approach
In May 2023, FDA began to consider the implications of AI/ML technologies for drug development with the publication of two discussion papers: Artificial Intelligence in Drug Manufacturing5 and Using Artificial Intelligence and Machine Learning in the Development of Drug and Biological Products.6 These two discussion papers highlight the agency’s areas of concern related to the incorporation of AI/ML in drug development and manufacturing.
Chief among these concerns are the governance, accountability, and transparency of AI/ML systems. For ML models, transparency and accountability are particularly challenging considering they are sub symbolic, or a “stack of equations — a thicket of often hard-to-interpret operations on numbers.”7 Thus, the nature of these systems makes its outputs difficult to interpret, presenting obvious regulatory challenges. To address these challenges, the FDA emphasizes the importance of “tracking and recording … key steps and decisions, including the rationale for any deviations and procedures that enable vigilant oversight and auditing.”8 The problem of transparency and accountability is further compounded by competitive concerns, as many of these models are proprietary.
Data quality is another concern the FDA addresses in its discussion papers, noting that the application of AI/ML systems in drug manufacturing can significantly increase the frequency and volume of data exchanges in the manufacturing process, thereby exponentially increasing the quantity of data. This increase in data output may require new considerations relating to data storage, retention, and security. In terms of data input, sponsors must be cognizant of any preexisting biases in the training data, as ML systems can easily duplicate or even amplify these biases.
The FDA also highlights reliability as another area of focus and concern. As recent experiences with large language models may attest, some AI systems are prone to hallucination, “a phenomenon where AI generates a convincing but completely made-up answer.”9 Indeed, in a recent study on AI hallucination, a group of researchers prompted a chatbot to generate a list of research proposals with reliable references. Of the 178 references provided by the chatbot, 69 did not have a digital object identifier (DOI), while 28 did not turn up on internet searches.10 Thus, FDA’s concern about reliability seems well founded, especially in the context of a drug development program.
The EMA’s Approach
Following the FDA’s recent publications, the EMA released a reflection paper11 advocating for a risk-based approach that considers patient safety and the reliability of development data. In April 2021, the European Union (EU) introduced a coordinated plan and a regulation proposal for AI, aimed at promoting innovation and ensuring AI benefits society. The reflection paper is an extension of this plan, outlining considerations for AI usage in drug development and emphasizing regulatory oversight based on risk assessment. It highlights three key concerns, specifically, the need for:12
- risk-based oversight,
- the establishment of strong governance for AI deployments, and
- guidelines covering data reliability, transparency, and patient monitoring.
The paper categorizes the risk of AI application in drug development stages. AI use in early drug discovery is deemed low risk, while its use in clinical trials spans various risk levels depending on factors like human oversight and potential impact on regulatory decisions. To manage risks, the paper recommends transparent AI models (the idea to fully trace information flow within a ML model), cautious handling of issues like overfitting (the result of non-optimal modeling practices wherein you learn details from training data that cannot be generalized to new data), and appropriate performance assessment metrics. Ethical and privacy issues, such as human agency and oversight, technical robustness and safety, privacy and data governance, transparency, accountability, societal and environmental well-being, diversity, non-discrimination, and fairness, are addressed and outlined.
Specific considerations for AI usage include ensuring accurate AI-generated text through quality review procedures and high-risk AI decisions in precision medicine settings, AI use in manufacturing adhering to quality risk management principles, and the importance of regulatory interactions during development. The reflection paper acknowledges that it is not an exhaustive source of regulatory insight on AI but serves as a starting point for further discussions. Stakeholders can provide feedback until Dec. 31, 2023.
Conclusion
While both the FDA and the EMA strive to provide a framework that balances innovation and patient safety, nuances emerge in their respective approaches. Stakeholder input and evolving industry practices are critical to shaping future regulatory guidelines. Collaboration among regulators, manufacturers, and researchers will be pivotal in fostering a transparent, accountable, and efficient AI ecosystem that enhances the development and deployment of medical products for the betterment of global health.
- Stanford University, “Artificial Intelligence and Life in 2030,” 2016, 12; https://ai10020201023.sites.stanford.edu/sites/g/files/sbiybj18871/files/media/file/ai100report
10032016fnl_singles.pdf. - FDA, “Using Artificial Intelligence and Machine Learning in the Development of Drug and Biological Products,” (May 2023), https://www.fda.gov/media/167973/download.
- Ibid.
- EMA, “5 Reflection paper on the use of Artificial Intelligence (AI) in 6 the medicinal product lifecycle.” 13 July 2023, https://www.ema.europa.eu/en/documents/scientific-guideline/draft-reflection-paper-use-artificial-intelligence-ai-medicinal-product-lifecycle_en.pdf.
- FDA, “Artificial Intelligence in Drug Manufacturing,” May 2023, https://www.fda.gov/media/165743/download.
- FDA, “Using Artificial Intelligence and Machine Learning in the Development of Drug and Biological Products,” May 2023, https://www.fda.gov/media/167973/download.
- Mitchell, Melanie. Artificial Intelligence: A Guide for Thinking Humans, p. 12.
- FDA, “Using Artificial Intelligence and Machine Learning in the Development of Drug and Biological Products,” p. 20.
- Athaluri SA, Manthena SV, Kesapragada VSRKM, Yarlagadda V, Dave T, Duddumpudi RTS. Exploring the Boundaries of Reality: Investigating the Phenomenon of Artificial Intelligence Hallucination in Scientific Writing Through ChatGPT References. Cureus. 2023 Apr 11;15(4):e37432. doi: 10.7759/cureus.37432. PMID: 37182055; PMCID: PMC10173677.
- Ibid.
- European Medicines Agency. (2023, July 13). Reflection Paper on the Use of Artificial Intelligence (AI) in the Medicinal Product Lifecycle. European Medicines Agency. https://www.ema.europa.eu/en/documents/scientific-guideline/draft-reflection-paper-use-artificial-intelligence-ai-medicinal-product-lifecycle_en.pdf.
- European Medicines Agency. (2021, August 16). Artificial Intelligence in Medicine Regulation. European Medicines Agency. https://www.ema.europa.eu/en/news/artificial-intelligence-medicine-regulation.