Greenleaf Regulatory Landscape Series
On February 4th 2020, the Secretary of Health and Human Services (HHS) declared that the threat to public health caused by the coronavirus justified the authorization of emergency use of in vitro diagnostics for the detection and/or diagnosis of COVID-19. This declaration, pursuant to section 564 of the Food Drug & Cosmetic Act (FD&C Act), allowed the Commissioner of the Food and Drug Administration (FDA or Agency) to issue Emergency Use Authorizations (EUAs) for developers of in vitro diagnostic tests aimed to detect and/or diagnosis COVID-19.
Following the declaration, the FDA began to receive a sudden flurry of EUA requests from diagnostic test developers before truly having an opportunity to develop its own guidance and recommendations for laboratories, manufacturers, and FDA staff. The FDA quickly developed guidance that outlined what specific tests required EUA authorization prior to marketing, what tests could use the notification pathway, and what validation and clinical testing was required for an EUA. The Agency focused on the authorization of molecular, antigen and antibody (serology) tests and informed healthcare providers and the public as to the type of information that each test could provide. As more became known about COVID-19 and as tests were in shortage across the United States, the FDA continued to evolve its regulatory framework and recommendations for in vitro diagnostic EUAs including publishing a revised guidance on its “Policy for Coronavirus Disease-2019 Tests During the Public Health Emergency.” Over the course of several months, the FDA published templates for both commercial manufacturers and laboratories for each of the three types of diagnostic tests and updated them with recommendations for the validation of pooled samples and the testing of asymptomatic individuals. The Agency also published templates for tests that use home specimen collection and for non-laboratory use tests.

Throughout the pandemic, the FDA has had to explore options to quickly allow diagnostic tests to reach patients while still ensuring that these tests are safe and effective. To enhance transparency around the FDA’s decision-making and to provide a forum for public questions, the FDA’s Center for Devices and Radiological Health (CDRH) began holding weekly virtual Town Halls for developers of diagnostic tests. These Town Hall meetings have been led by Dr. Timothy Stenzel and Toby Lowe from CDRH’s Office of In Vitro Diagnostics and Radiological Health. Each week, CDRH staff provides developers with an update on any major changes in the FDA’s policies related to diagnostic tests and provide insight on what changes may be coming in the near future. Dr. Stenzel has also expressed the Agency’s willingness to work with developers of novel technologies and to remain flexible in its regulatory approaches. CDRH has already held 30 Town Hall meetings for diagnostic test developers and has committed to holding these events throughout October 2020.
During the Town Hall meetings, Dr. Stenzel shared that the FDA has received more than 2,000 EUA requests from developers and explained the triage process that CDRH has developed in order to respond to these requests with the resources that the Agency has available. Recently, Dr. Stenzel noted that after issuing more than 200 EUAs for diagnostic tests, the FDA is now prioritizing EUA requests for specific types of tests including point-of-care tests, at-home tests, and tests with high-throughput capacity with ability to scale. He explained that the FDA is still taking EUA requests for tests that do not have these specific features but that they will receive lower priority in terms of review. Another major development that was recently announced during a CDRH Town Hall meeting is that CDRH will no longer be reviewing EUAs for Laboratory Developed Tests (LDTs). This comes after the Department of Health and Humans Services (HHS) published a notice in August stating that the FDA would no longer require premarket review of LDTs for COVID-19. Although HHS’s announcement stated that companies could still voluntarily submit EUA requests for LDTs, the Agency has since stated that it will no longer review them. Dr. Stenzel explained that this change was made to help focus the FDA’s resources on the EUAs that have been identified as priority. During last week’s Town Hall meeting, Dr. Stenzel noted that CDRH will soon be updating their FAQ pages to provide further clarity on how this policy will impact current LDT submissions and EUAs that are already authorized.
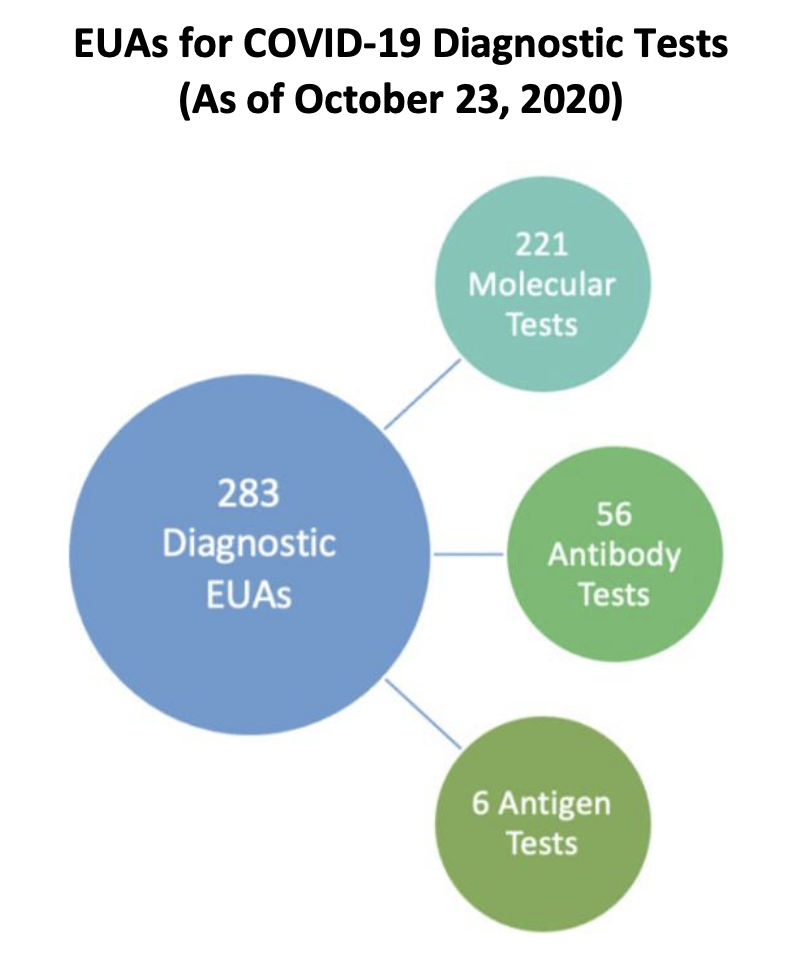
As of October 23, 2020, there are 283 EUAs that are authorized for COVID-19 diagnostic tests
including 221 molecular, 56 antibody and 6 antigen tests. FDA’s website also provides a complete list of all current EUA authorizations for the following EUA categories:
- Individual EUAs for Molecular Diagnostic Tests for SARS-CoV-2
- Umbrella EUA for Molecular Diagnostic Tests for SARS-CoV-2 Developed And Performed By Laboratories Certified Under CLIA To Perform High Complexity Tests
- Individual EUAs for Antigen Diagnostic Tests for SARS-CoV-2
- Individual EUAs for Serology Tests for SARS-CoV-2
- Umbrella EUA for Independently Validated Serology Tests for SARS-CoV-2
- Individual EUAs for IVDs for Management of COVID-19 Patients
Overall, the FDA has made significant progress in authorizing EUAs for COVID-19 diagnostic tests and has helped to facilitate access to these tests for patients across the United States. FDA’s regulatory policies will continue to be updated as the pandemic changes and as developers begin to transition their EUA authorizations to full premarket clearance or approval.
Related Sources
- Coronavirus Disease 2019 (COVID-19) Emergency Use Authorizations for Medical Devices
- Policy for Coronavirus Disease-2019 Tests During the Public Health Emergency (Revised)
- Virtual Town Hall Series – Coronavirus (COVID-19) Test Development and Validation
- Rescission of Guidances and Other Informal Issuances Concerning Premarket Review of
Laboratory Developed Tests - In Vitro Diagnostics EUAs
- Coronavirus (COVID-19) Update: Daily Roundup October 23, 2020
- A Closer Look at the FDA’s Center for Devices and Radiological Health’s Unprecedented Efforts in
the COVID-19 Response